
Lindy
Lindy 3.0 的发布,标志着 AI 智能体进入了一个新阶段:创建更简单,一句话生成可用智能体;能力更完整,摆脱 API 限制,操作任何系统;协作更高效,团队共享、统一管理、规模化落地。它让我们离“AI 员工”的愿景更近了一步——不是替代人类,而是扩展团队的能力边界。
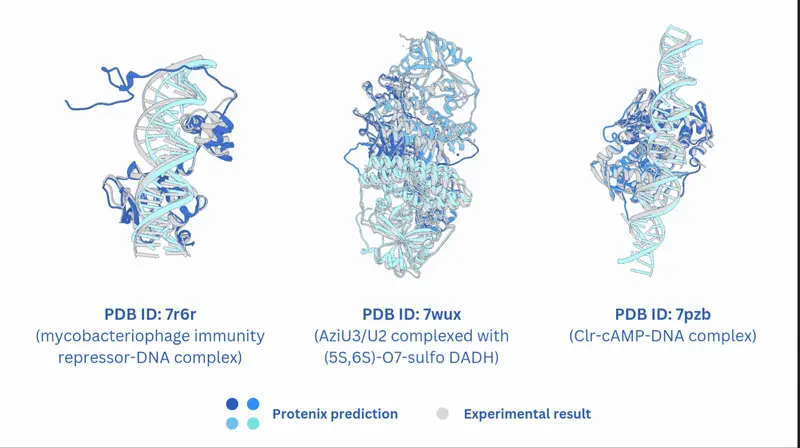
Protenix 是面向高精度生物分子结构预测构建的开源体系,也是字节跳动 Seed 团队在计算生物学领域,为推动开放、可复现、可扩展研究工具迈出的关键一步。Protenix-v1 是首个在严格对等条件下性能超越 AlphaFold 3 的全开源生物分子结构预测模型,支持蛋白质、核酸、配体等多类型分子的通用结构预测,不仅开放模型权重,同时...
在计算生物学领域,一个长期存在的难题终于迎来突破:字节跳动 Seed 团队开源的 Protenix-v1,成为首个在严格对等条件下性能达到甚至超越 AlphaFold 3 的完全开源模型。
这意味着,科研人员、药物开发者和开源社区,终于拥有了一个透明、可复现、高性能的工具,用于预测蛋白质、核酸、配体及其复合物的三维结构——而无需依赖闭源黑箱。
AlphaFold 3 虽然强大,但其闭源性质限制了科学界的深度验证、定制与改进。此前的开源模型(如 Boltz-1、Chai-1)虽有进展,但在关键任务(如抗体-抗原结合预测)上仍存在显著差距。
Protenix-v1 不仅填补了这一空白,更展现出一项独特能力:推理时扩展(Inference-Time Scaling)——通过增加采样数量,预测精度可持续提升,为实际应用提供了明确的“计算成本 vs. 精度”权衡机制。
例如,在抗体-抗原复合物预测中,将采样种子从 5 个增至 80 个,DockQ 成功率从 36% 提升至 47.7%,这是此前开源模型无法实现的。
Protenix 支持多种生物分子的通用结构预测:
所有预测均输出 3D 坐标 + 置信度分数(pLDDT、pTM、ipTM),便于后续筛选与实验验证。
在修正后的 FoldBench 和自建 PXM 基准集上,Protenix-v1 表现如下:
| 任务类型 | Protenix-v1 | AlphaFold 3 | 优势 |
|---|---|---|---|
| 抗体-抗原 | 52.31 (DockQ SR) | 48.75 | +3.56% |
| 蛋白质-RNA | 68.46 | 65.22 | 显著领先 |
| 蛋白质-配体 | 62.54 | 62.59 | 基本持平 |
| 蛋白质单体 | 0.8857 (lDDT) | 0.8803 | 略优 |
注:所有测试均在相同数据截止日期(2021-09-30)、模型规模与计算预算下进行,确保公平比较。
更令人振奋的是其实用增强版 Protenix-v1-20250630:在引入更新训练数据后,抗体-抗原预测成功率在 PXM-2024 上跃升至 64.02%,展现了强大的可扩展性。
安装与预测极其简单:
# 安装
pip install protenix
# 预测(JSON 输入)
protenix pred -i input.json -o ./output -n protenix_base_default_v1.0.0
项目提供详细 inference_demo.sh 示例,涵盖多分子输入格式、模板使用、置信度过滤等场景。
对于高精度需求(如临床前候选),建议启用 80+ 种子采样;对于高通量筛选,标准 5 种子配置即可平衡效率与准确性。













